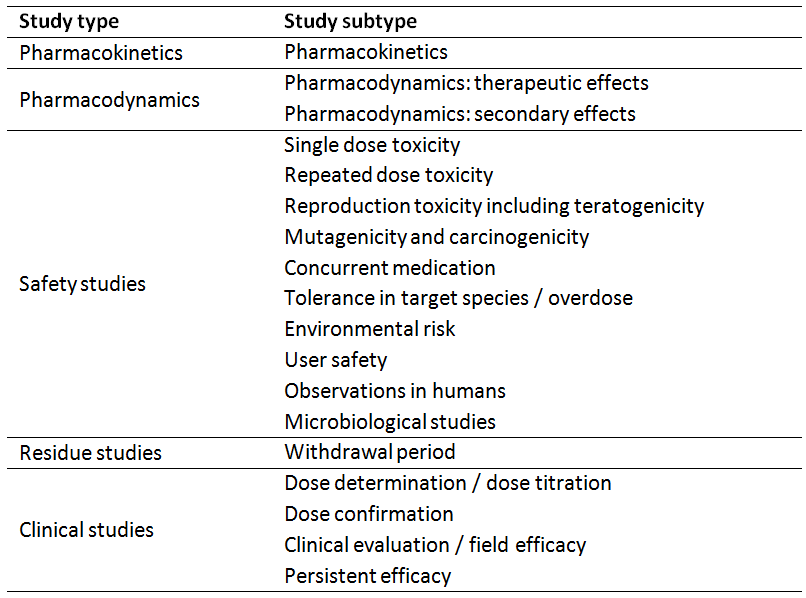
Table 1: Relevant studies to demonstrate efficacy and safety of a VMP (European Commission, 2015)
Development of a Database for Study Data in Registration Applications for Veterinary Medicinal Products
Anke Finnah1*
1Federal Office for Consumer Protection and Food Safety (BVL), Berlin, Germany
2Department of Biometry, Epidemiology and Information Processing, WHO Collaborating Centre for Research and Training in Veterinary Public Health, University of Veterinary Medicine, Hannover, Germany
*Corresponding Author (anke.finnah@bvl.bund.de)
Vol 2, Issue 1 (2017)
Published: 09 Feb 2017
Reviewed by: Laura Urdes (PhD, DVM, PgDip, CertAqV) and Peter Rodgers
DOI: 10.18849/VE.V2I1.49
Objective: In the present study, the feasibility of a systematic record of clinical study data from marketing authorisation applications for veterinary medicinal products (VMP) and benefits of the selected approach were investigated.
Background: Drug registration dossiers for veterinary medicinal products contain extensive data from drug studies, which are not easily accessible to assessors.
Evidentiary value: Fast access to these data including specific search tools could facilitate a meaningful use of the data and allow assessors for comparison of test and studies from different dossiers.
Methods: First, pivotal test parameters and their mutual relationships were identified. Second, a data model was developed and implemented in a relational database management system, including a data entry form and various reports for database searches. Compilation of study data in the database was demonstrated using all available clinical studies involving VMPs containing the anthelmintic drug Praziquantel.
By means of descriptive data analysis possibilities of data evaluation including graphical presentation were shown. Suitability of the database to support the performance of meta-analyses was tentatively validated.
Results: The data model was designed to cover the specific requirements arising from study data. A total of 308 clinical studies related to 95 VMPs containing Praziquantel (single agent and combination drugs) was selected for prototype testing. The relevant data extracted from these studies were appropriately structured and shown to be basically suitable for descriptive data analyses as well as for meta-analyses.
Conclusion: The database-supported collection of study data would provide users with easy access to the continuously increasing pool of scientific information held by competent authorities. It enables specific data analyses. Database design allows expanding the data model to all types of studies and classes of drugs registered in veterinary medicine. The needs for detailed data recording and versatility of the data model must be carefully balanced.
Application: The database will be used by regulatory authorities.
Medicinal products for human or veterinary use need to have suitable properties in terms of quality, safety and efficacy, which must be shown by scientific studies and other scientific literature. The supporting documents are compiled in registration dossiers, which are the primary source of information for evaluating medicinal products by regulatory authorities.
In Germany, veterinary medicinal products (VMPs) are authorised by the Federal Office for Consumer Protection and Food Safety (Bundesamt für Verbraucherschutz und Lebensmittelsicherheit, BVL). This authority keeps a large amount of scientific documentation related to VMPs. Registration dossiers usually consist of four parts, of which Part III Safety and Part IV Efficacy contain the necessary information including study reports on pharmacology, toxicology, safety, and efficacy of the product or active compound. Table 1 gives an overview of the main studies, which must be submitted along with the application for authorisation.
The authority has archived all applications for authorisation including the supporting dossiers in electronic or in paper form. Administrative product data are managed in a central database (Arzneimittel-Informationssystem, AMIS), which contains the relevant administrative product data (including Summary of Product Characteristics, SPCs) and is used for management purposes and to provide (non-confidential data) information to the public. Scientific data from registration dossiers have not been stored in a database so far. Therefore, if assessors need to make use of study data, they have to extract them from the paper or electronic dossiers.
In human medicine, various databases for the registration of clinical studies on the efficacy of pharmaceuticals have been established in recent years (ISRCTN Registry, 2016; ClinicalTrials.gov, 2015; German Clinical Trials Register, 2014). Based on the Declaration of Helsinki on Ethical Principles in Medical Research in Humans (World Medical Association, 2013), clinical trials must be registered in publicly available databases and the results of these trials must be published, irrespective of their outcome.
In the field of regulation of human medicinal products, the European Medicines Agency established a European Clinical Trials Database (EudraCT, 2016). EudraCT is used by national regulatory authorities to support the supervision of clinical trials. It was established as a confidential database, in accordance with article 11 of Directive 2001/20/EC. Some of the information held in the EudraCT database has also become publicly available (EU Clinical Trials Register, 2016).
In veterinary medicine no similar database systems for clinical studies have been established so far.
In 2015 an international consortium started the VetAllTrials initiative, dedicated to the development of one or more veterinary clinical trial registries (VetAllTrials, 2015). Some attempts at collecting study data in veterinary medicine with limited focus and user groups have been made (e.g. HomVetCR: Clinical Research on Veterinary Homeopathy, 2011; Coordination Staff for Veterinary Clinical Studies, n.d.). In the absence of databases for systematic collection of data from studies in veterinary medicine, up to now only publication databases like Medline or Veterinary Science Database have been able to be used.
In accordance with the approach in practice for studies on human medicinal products, also for registration studies on veterinary medicinal products, a database for authority use should be established. Decisions whether parts of it may become publicly available should be reached at a later time.
Nowadays the majority of marketing authorisations for VMPs in the European Union are granted by way of decentralised (DCP) or mutual recognition procedure (MRP) involving more than one Member State (MS). This is closely followed by an increasing exchange of information between competent authorities (CA) in order to come to a consensual decision. Moreover, new scientific findings, new safety risks and other issues may trigger a Union Referral Procedure as laid down in Articles 33, 34, 35, and 78 of Directive 2001/82/EC, Article 45 of Regulation (EC) No. 728/2004 or Article 13 of Commission Regulation (EC) No. 1234/2008. In these referrals, an assessment is made by the network and an opinion given on the questions raised. Normally, the assessment is based on answers provided by concerned marketing authorization holders (MAHs), supported by data from the original registration dossiers. Thereby, known studies are re-evaluated in consideration of the new aspects.
It is obvious that a common database, which includes all studies submitted to European CAs, would be a useful tool to smoothen all these procedures within the Network.
The objective of this feasibility study was to develop a database for recording data from drug registration studies. The pivotal steps of this study included the construction of a relational data model, the implementation of the data model in a database, and the testing of functions and application options of the database. Finally, application examples from an authority perspective were presented.
Data source
The dossiers for registration applications for VMPs submitted to BVL between 1974 and 2010 served as the data source. From the total number of dossiers a subset of 95 was selected encompassing all applications for VMPs containing the anthelminthic agent Praziquantel as either the only or one of several active substance(s). Data from Part I and Part IV of the dossiers, i.e. data concerning the drugs and data on clinical efficacy studies were included in this feasibility study. Within this publication, no confidential data are disclosed.
Data model design and implementation in a prototype database (MS Access)
Data modelling was based on the entity-relationship method (Chen, 1976). First, the dossiers were screened and the main product data and relevant study data were identified. Entities, attributes and their relationships were determined as the key components of a relational data model.
Rules of normalisation were applied to the entire study and product data. To this end, all tables were split to remove any data redundancy and to resolve dependencies between non-key fields (Klug, 2008).
The data model was constructed of data tables, assignment tables and lookup tables. Each table contained an attribute which served as a primary key that uniquely identified a data set. Primary keys were labelled with the suffix “ID” (e. g. Study_ID, Result_ID).
Each data table represented an entity including its attributes. Lookup tables provided selection lists of terms to the user (e. g. list of species or active substances). Some of these lists were based on internationally agreed terminology in the field of drug registration and pharmacovigilance (ATCvet, 2016; EDQM Standard Terms, 2016).
Tables were linked by 1:n relationship and the primary key of the reference table served as foreign key in the associated tables (Bildner, 2008).
The relational data model was implemented in a prototype database using MS Access 2003.
Statistical evaluation
Following transfer of the data from Access to SAS (Version 9.1.3, Statistical Analysis System, SAS Institute, Cary, NC, USA) descriptive data analyses were performed.
If applicable, significance testing was applied using the Mann-Whitney U test and the Kruskal-Wallis test (Clauß and Ebner, 1989). The null hypothesis of no differences between groups under observation was rejected if the p-value was below 5%.
Data source: choice of compound and study types
In veterinary medicine, the most frequently used medicinal products are indicated for the treatment of parasitic and bacterial infections and the majority of authorised VMPs in Germany as well as in the European Union are antiparasitics and antibiotics (IFAH Annual Report, 2013).
Compounds considered suitable for the purpose of this feasibility study should already have been on the market in Germany for a long time (since the intention was to include product and study data from the early days of German Drug Law until the deadline in 12/2010). The products should have been approved for use in food-producing animal species and pet animal species. They should have been authorised by national, decentralised / mutual recognition or centralised procedure to cover the different data requirements involved. These criteria were best met by the antheminthic compound Praziquantel. This agent is used in dogs, cats, horses and sheep to treat infestations with flatworms (Plathelminthes) like tapeworms and trematoda. It is available as single agent VMP or combination VMP, i.e. in combination with other anthelminthic agents.
Three subtypes of clinical studies on the efficacy of Praziquantel were identified in the dossiers, namely dose determination/dose titration studies, dose confirmation studies, and clinical evaluation/field studies.
All studies meeting the criteria listed in Table 2 were included in the database prototype and subsequently used for data analysis.

Data model design
The relational data model was based on the two pillars product data, i.e. administrative data and product characteristics, illustrated by the central entity Data_Product and data from clinical studies illustrated by the central entity Data_StudyAdminData. Product data and study data were linked through an n:m relationship to ensure multiple possibilities of interaction between both.
An assignment table (Zuord_StudyAdminData_Product) was placed between these central entities in order to break down their relationship into two 1:n relationships.
The following sections give a brief description of the entities, their attributes and relationships resulting from the requirements analysis and implemented in the data model. An overview of the complete relational data model is given in Figure 1. Terms referring to those used in the data model are printed in italics.
Product data
Product data were mapped in the data model by the central entity Data_Product. This entity contains attributes that are commonly used in the daily work of regulatory authorities and the pharmaceutical industry. These are: (1) name of the drug (VMP); (2) submission number (ENR) which is a unique seven-digit number allocated to each VMP for which registration application has ever been submitted; (3) authorisation number (AuthorNumb) which is a unique number allocated to approved products; (4) dosage form (DosForm) such as tablet, solution, ointment; (5) ATCvet code (ATCcode) based on the ATC index for the classification of drugs (ATCvet, 2016); and (6) the authorisation procedure (Procedure) which is either national, decentralised or centralised depending on the European Member States involved. Four other entities were connected with the central entity. They provide selection lists (Prf) of active substances, dosage forms, ATCvet codes, and application procedures (Prf_Substance; Prf_DosageForm; Prf_ATC; Prf_Procedure). From these, the entity Prf_Substance was linked via assignment table (Zuord_Product_Substance) to the central entity since VMPs may be composed of more than one active substance and each active substance can be used in several products.
Study data
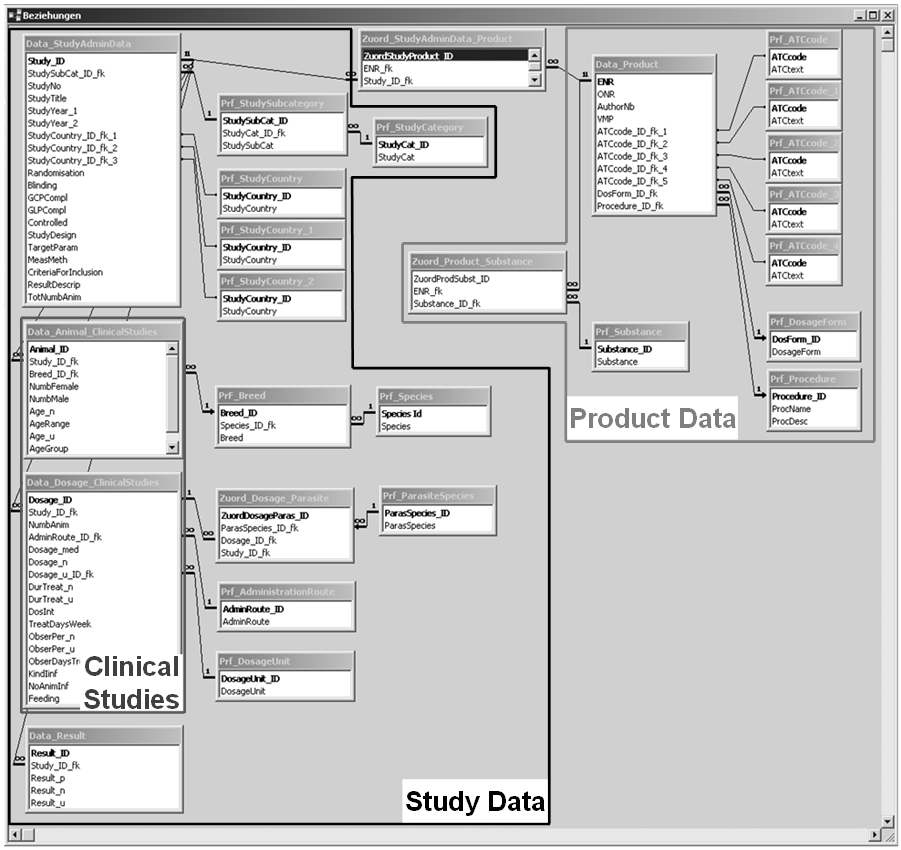
The other part of the data model illustrates the relevant study-related data and the relationships thereof. Data_StudyAdminData which encompasses attributes of purely administrative nature and attributes which focus on the study design constitutes the central entity. These attributes are: (1) the type of the study (StudySubCat) as shown in Table 1; (2) the study identification number (StudyNo) which was allocated by the sponsor; (3) name of the study (StudyTitle); (4) beginning and end of the study (StudyYear1, StudyYear2); (5) place of study (StudyCountry1, StudyCountry2 etc.); (6) - (10) randomisation of the test animals; blinding of the assessors; compliance with Good Clinical Practice (VICH GL 9, 2000); compliance with Good Laboratory Practice (Directive 2004/9/EC, Directive 2004/10/EC); and the inclusion of a control group. Combo boxes (Yes/No/Unknown) were added for the attributes (6) to (10). Data entries of the attribute study type and place of study were made by means of look-up tables. Text fields were added for (11) the description of the study design (StudyDesign); (12) object of the study (TargetParam); (13) methods used (MeasMeth); (14) inclusion and exclusion criteria (CriteriaForInclusion); and the description of the results (ResultsDescript). Lastly, (15) the total number of animals (TotNumbAnim) was included in the central entity.
Another important entity which is linked through a 1:n relationship with the entity Data_StudyAdminData compiles the quantitative study results (Data_Result). Irrespective of the type of study, all types share the central entity and the entity Data_Result.
Two specific entities were added with data typical for clinical studies: The entity Data_Animal_ClinicalStudies contains attributes related to the test and control animals, such as number of male and female individuals (NumbMale, NumbFemale), species and breed. The other entity Data_Dosage_ClinicalStudies presents pivotal data on treatment regimen, such as number of animals per dosage group (NumbAnim) and information on drug(s) and dosage (Dosage_med, Dosage_n, Dosage_u_ID_fk).
If the data model is made applicable to other types of studies (non-clinical studies) it is easy to achieve by adding further entities to the basic structure. Thus, extensions can be made without the need for modifications to the original data model.
Implementation in MS Access
Microsoft Access 2003 was used to implement the data model (Figure 1).
Additional features were implemented to allow for convenient input of study data and also for database queries with data outputs in a comprehensive and clear format.
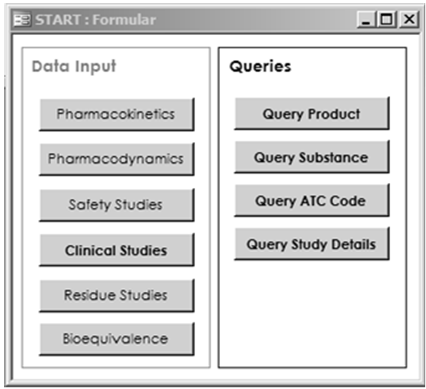
The main functions for database users are data entry and search functions. Convenient access to these functionalities was provided by a top menu (Figure 2), which leads to data entry on the one hand and to queries on the other hand. The items printed in bold have already been realised in the prototype; these may be supplemented in an extended version.
For data entry, a form which automatically fills the data tables and includes drop down lists for contents of lookup tables, was created. Besides ease of use, this helps to avoid errors in data entry as data sets in all related tables are generated automatically.
Four queries were implemented which exemplify ad-hoc searches on study data. These queries aim to identify those studies available per product, per active substance and per ATCvet code. Main study data are given as tabular outputs. Complete details of study reports can be requested with the fourth query on study details.
These basic search functions may be extended by the addition of filters in further database versions. In reports a) to c) filters for study year, target species or study quality (randomisation, blinding, and number of animals included) would produce more specific results, provided that a sufficient number of studies are in the database. The use of filters should be optional. Users are able to obtain an overview of the available studies and then select an appropriate subset of studies.
Descriptive data analysis of selected studies
Descriptive statistical analyses of study data demonstrate what additional information can be obtained if all clinical studies archived in the BVL are available in a database system.
Due to the feasibility nature of the study, results shown below may serve as examples only and do not allow conclusions to be drawn concerning the entire amount of clinical studies submitted to BVL. As only a small subset of the available study data was included in the database prototype, results should not be interpreted regarding their contents but rather the possibilities for data usage thereof.
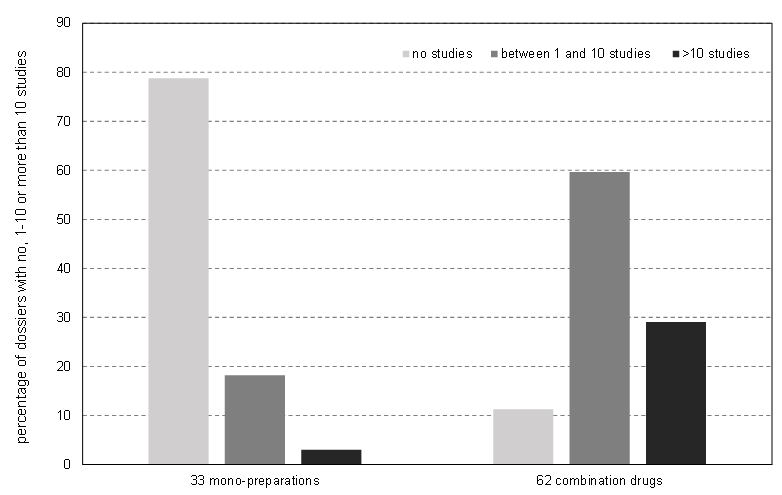
Data from 308 clinical studies derived from registration dossiers of 95 veterinary drugs containing Praziquantel (from which 33 were single agent products and 62 combination products) met the inclusion criteria (Table 2). Data from these clinical studies were largely similar and could be recorded in a structured manner and were therefore suitable to be included in the database.
The dossiers of 33 products (=35%) contained bibliographical data or were referenced to other authorised drugs. Therefore, no clinical studies were presented in the dossiers of these products.
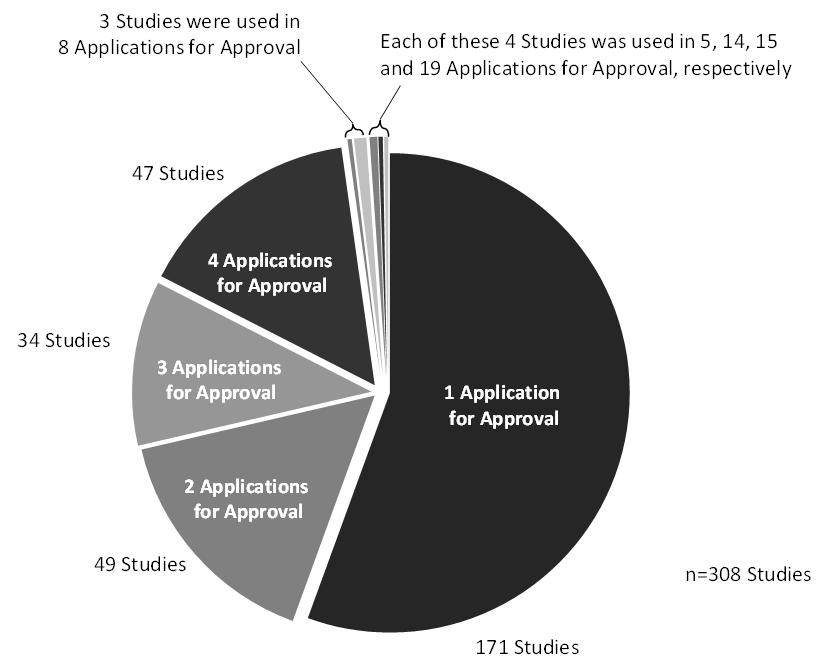
Seventy-five percent of the dossiers contained between 0 and 8 studies, with 35% containing no studies and 40% containing between 1 and 8 studies. The remaining dossiers had between 9 and 100 studies. The maximum of 100 studies was found in one dossier from 1993. As most of these studies were not conducted and presented according to current guidelines, with very small numbers of animals per study, they would not be counted as full-value studies nowadays. See Figure 3.
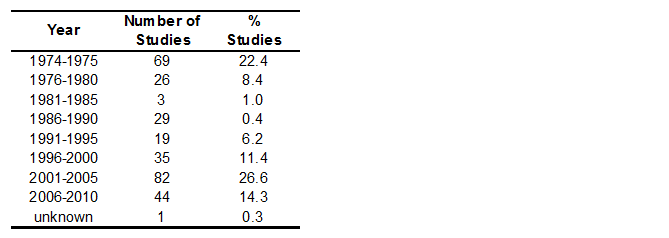
Stratification of number of studies per 5-year period gives information on distribution of years studies were conducted, which has to be considered when assessing changes of certain parameters over time. Up to 1975, already 69 studies on the single agent products had been conducted; further studies on different combinations of Praziquantel with other substances have been conducted in recent years. As in all periods studies were carried out, study evaluation and time series analysis with regard to quality-related parameters is possible (see Table 3).
Several other analyses were performed, looking at the number of studies used in one or several marketing authorisation applications (Figure 4) or vice versa on the number of studies per dossier, which gives information on the re-use of studies for more than one drug and on the amount of clinical data per dossier. Re-using studies in more than one dossier is possible if a company applies for or is granted more than one marketing authorisation for the same or a similar product.
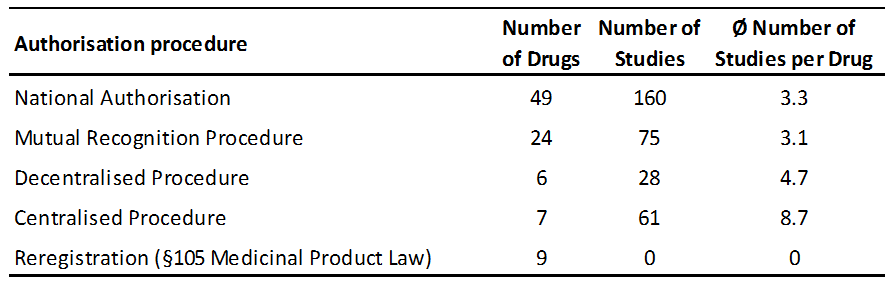
The total number of animals included in a clinical study is an important criterion for the power of the study and herewith its quality. In the selected subset of studies, study size was between one (only in very old and pilot studies) and 539 animals per study (median 20 animals). Based on the limited amount of studies included further stratifications were performed for study size in different kinds of authorisation procedures. Further stratifications would be interesting to look at in larger numbers of clinical studies, e.g. per species or study subtype.
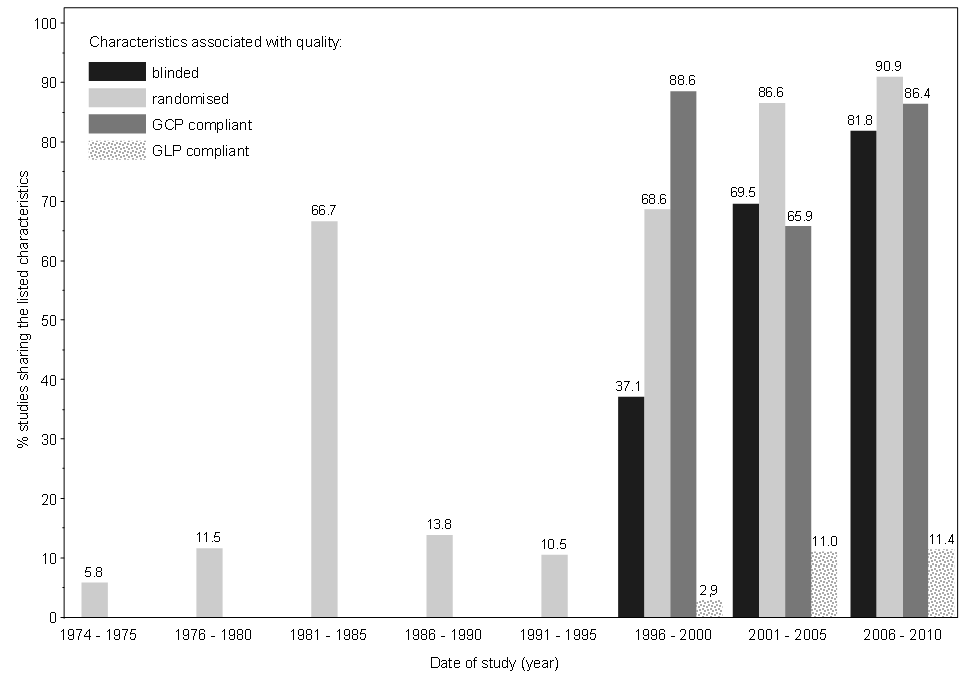
Some parameters associated with quality of studies were analysed for variation over time. This may be interpreted against the background of changes in legislation and adoption of guidelines for conducting clinical studies. Whereas the study size, measured as numbers of animals per study, did not significantly increase, the proportion of studies with blinding, randomisation, GCP and/or GLP compliance clearly increased over time (Figure 5). For a larger amount of studies, this analysis should be performed not for time intervals but for single years to see if implementation of guidelines have led to greater changes in study quality.
Evaluation of the data model and critical assessment of study data quality
The data model developed in this feasibility study has proved to be suitable for presenting clinical study data from registration dossiers. It contains the most relevant information on veterinary medicinal products and their active substances. For the study data, a modular approach has been chosen, consisting of more general data relevant for all study types (administrative data, e.g. study number, date, title, results) and of data with specific information applicable for one study type only. Information from clinical studies has been used as an example here, but tables for other study types can be added without any need for changes to the already existing structure.
No major flaws were noticed during practical testing. However, it became evident that some minor changes to the data model would clearly improve performance and usability of the database.
In one aspect the data model should even undergo some simplification: Data table Data_Dosage_ClinicalStudies contains information on the parasite species a drug is effective against, which is applicable for products for antiparasitic use only. Knowledge gained on the current study population showed that parasite species were the same among tested dosage groups. Thus, combining dosage group and parasite species is considered unnecessary. As the data model should also be suitable for clinical studies on other kinds of drugs, the demands of detailed recording of data should be balanced with the need for versatility of the data model. Therefore, information concerning the parasite species should be integrated in the more general target parameter data field (data field TargetParam in table Data_StudyAdminData). Otherwise, inclusion of different data tables for different classes of drugs would lead to an unreasonably more complex data model.
For use of a study database in live operation, constant updating of product data would be necessary. Therefore, an interface between study database and product database would have to be implemented.
As shown in the descriptive data analysis, the quality of studies, as well as the quality of study reports have improved in more recent clinical studies. In the set of studies used for testing, problems in data recording due to insufficient or missing data were found in older studies only.
Examples of problems in data recording due to shortcomings in data quality and presentation of data are missing study numbers or inconsistencies regarding contents such as differences in numbers of parasites under consideration between study plans and final study reports. As only indications based on endpoints predetermined in study plans would be accepted, this may have an impact on the outcome of authorisation procedures. Missing values were found concerning numbers of animals per treatment group. Also, in some older studies, only summary reports were available with missing information on aspects like inclusion or exclusion criteria. In any case, entry of data should be combined with a check of data quality and it may be useful to have a text field in the administrative part of the data mentioning problems in data quality.
Outlook
Tabulated study reports
In this feasibility study, complete study reports were browsed for those data considered important to be stored in the database prototype. This time-consuming work would be reduced significantly for further studies if these particular data were made available in an easy-to-view format. As already available for toxicological studies, also for clinical studies uniform templates are considered useful to provide a quick overview of studies regarding protocol- and results-related information. It would also avoid burdensome extraction of data from study reports and facilitate the process of data entry into a study database.
The authors therefore suggest that all studies in application documents of veterinary medicinal products should provide tabulated study reports containing the parameters used in the database prototype. Based on this initial proposal, involved stakeholders, the pharmaceutical industry and regulatory authorities would need to further discuss and agree on a set of relevant parameters to be included in a template for tabulated study reports. An appropriate template should become part of the “Notice to Applicants” documents.
Potential uses and benefits of a study database
The majority of studies submitted to BVL are in support of drug registration applications. These studies are assessed within the application process.
For post-authorisation surveillance of safety or efficacy, assessors might seek to gain further information from studies conducted with similar products. Moreover, the exchange of scientific information has attained greater importance due to cooperation within the network of European regulatory authorities. Summaries of study reports are commonly requested from the national authorities that granted the first marketing authorisation of a medicinal product in a European member state. In addition, for streamlining products by means of harmonisation procedure (e.g. indications or withdrawal periods) compilations of available study data are needed.
A study database may be used to gain an overview of all available study data on for example one specific substance, either to have a summary of the current level of knowledge or to conduct meta-analysis to acquire a common estimate for the endpoint of interest.
At present, it is cumbersome to select the relevant studies from individual dossiers. Doubtlessly, this would be accelerated by saving all incoming studies in a database. Such a knowledge database would serve for secondary use of data by providing elaborated search functions in combination with direct access to study data.
Considerations regarding a study database would have to address retrospective inclusion of study data. This feasibility study revealed large differences in the quality of clinical studies and of study reports, with clear tendency towards higher quality in more recent clinical studies, while early studies often failed to meet present-day requirements. Thus, the cost-benefit-ratio for including older studies might not be balanced and the authors would recommend limiting data collection to studies from a given cut-off point onwards. A reasonable starting point could be the year 2001, when the relevant legislation (Directive 2001/82/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to veterinary medicinal products) came into effect. Alternatively, starting points could be set for groups of drugs or active substances dependent on the date the relevant guideline documents on conducting studies came into effect.
Further perspectives
In the long term, it would be reasonable to establish a system for direct transmission of electronic study-related data into suitable databases accessible to regulatory authorities. For this purpose, technical standards concerning data format and structure of data need to be developed. It lies within legislation to pave the way forward.
Acknowledgments: The authors would like to thank Stephan Steuber for his helpful contribution to the sighting of VMPs for this research project.
Competing interests: None to declare. The manuscript, or parts of it, have not been and will not be submitted elsewhere for publication. None of the authors have any financial or personal relationships that could inappropriately influence or bias the content of the paper.
Intellectual Property Rights
Authors of articles submitted to RCVS Knowledge for publication will retain copyright in their work, but will be required to grant to RCVS Knowledge an exclusive licence of the rights of copyright in the materials including but not limited to the right to publish, re-publish, transmit, sell, distribute and otherwise use the materials in all languages and all media throughout the world, and to licence or permit others to do so.
Authors will be required to complete a licence for publication form, and will in return retain certain rights as detailed on the form.